【與時間競賽】2個月大BB患罕見病需注射全球最貴藥物 父母為救愛兒短期籌1300萬港幣
人間有愛!新加坡一名2個月大的男嬰自出世就患上罕見病「脊髓性肌肉萎縮症」,急需240萬新加坡幣(逾1300萬港幣)的藥物費用進行治療。然而,父母不願放棄,故於網上發起眾籌,卻意外獲大量網民支持,並在短時間內籌夠所需費用。所幸的是,現在男嬰已在醫院完成治療,也能回家休養,情況良好。
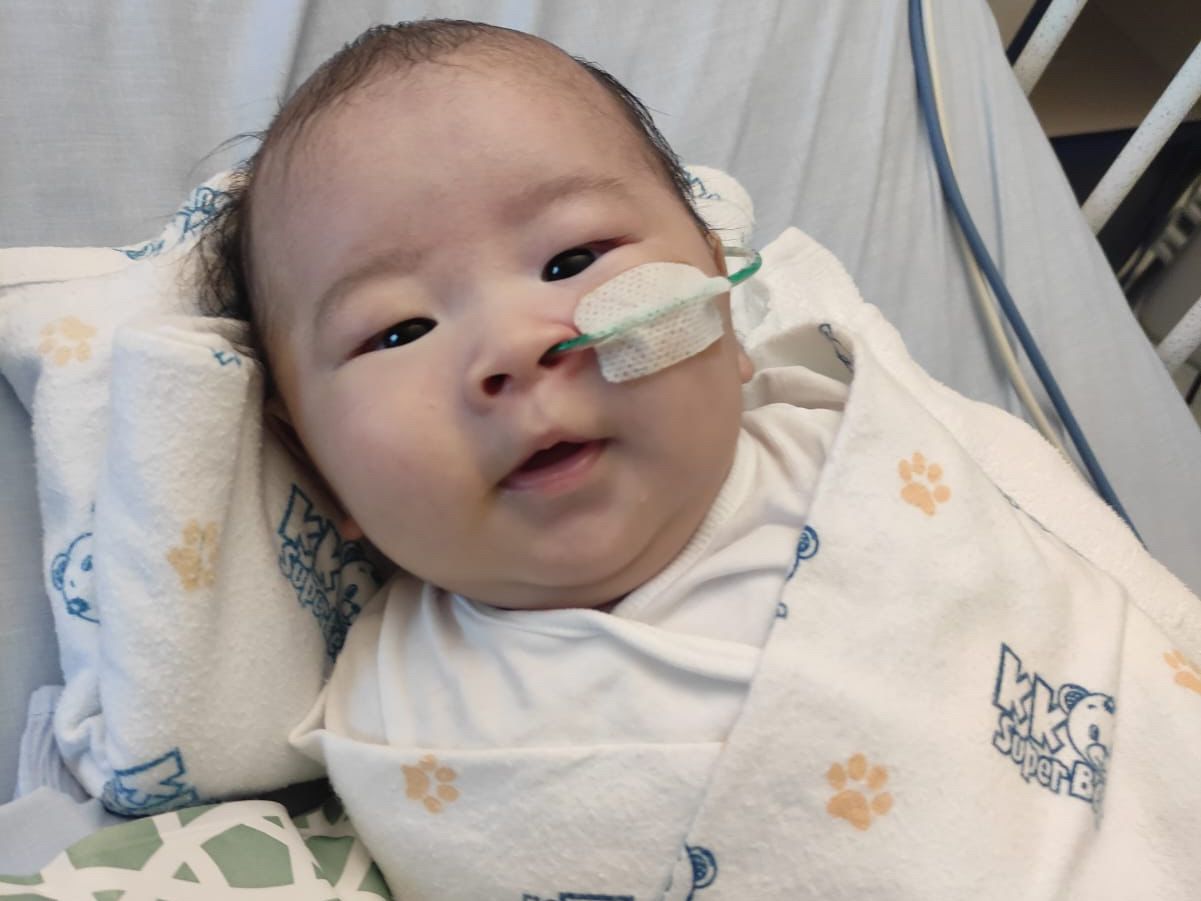 Photo from hope_for_baby_lucas IG
Photo from hope_for_baby_lucas IG
BB出世發現異常症狀
綜合外媒報導,新加坡43歲男子王梓全與其40歲的妻子綺恩結婚10年,經歷了2次的人工受孕後,終於在今年的4月迎來了他們的第一個BB王彥喆。然而,當時王彥喆出世後,卻發現雙腿失去活動能力,而且頸部也無法支撐重量,導致頭部經常側向一邊。
發燒檢查揭患罕見病
直到6月初,王彥喆因突然發高燒,父母於是帶他去醫院進行詳細檢查,才證實他患上罕見病「脊髓性肌肉萎縮症」。雪上加霜的是,當檢查出患上此疾病時,情況已經相當嚴重,由於呼吸與吞嚥肌肉已受到影響,因此他需要通過插管進食與服用藥物,而且還需要通過檢測器來監察血氧與心跳,甚至連呼吸都需要儀器輔助。
治療藥物需高達240萬新加坡幣
對此,夫婦倆感到絕望與無助,但醫生表示有一種藥物,是由美國食品與藥物管理局(FDA)在2019年批准的一種名為Zolgensma的藥物,通過替換缺失或不起作用的運動神經元存活基因的功能,可抑制病情惡化,阻止進一步損害肌肉並維持生存所需的剩餘肌肉功能,治療預期效果良好。然而,其藥物費用卻在每次注射要高達240萬新加坡幣(1300萬港幣),同時也被譽為全世界最貴的藥物,更是沒有得到政府的補貼,因此患者需要獨立支付藥物資金。
 Photo from hope_for_baby_lucas IG
Photo from hope_for_baby_lucas IG
父母:盡力給我們的兒子一個生存的機會
雖然這個高昂費對於夫婦倆來説是一個難以承擔的數額,但他們不放棄,並表示「作為父母,我們必須盡一切努力給我們的兒子一個生活的機會」於是就在社交媒體上發起衆籌,希望大家可以幫助他的兒子。對此,母親表示,「隨著他的肌肉每一秒都在惡化,在造成更多傷害之前為他提供治療,就像是一場與時間的賽跑」。但所幸,由於網絡的快速傳播,王彥喆的故事也迅速的引起了網民的關注與同情並紛紛捐款,然而卻在短短的12日裡共籌得了240萬的新加坡幣,而這筆款項也已經足以支付王彥喆的藥物費用。
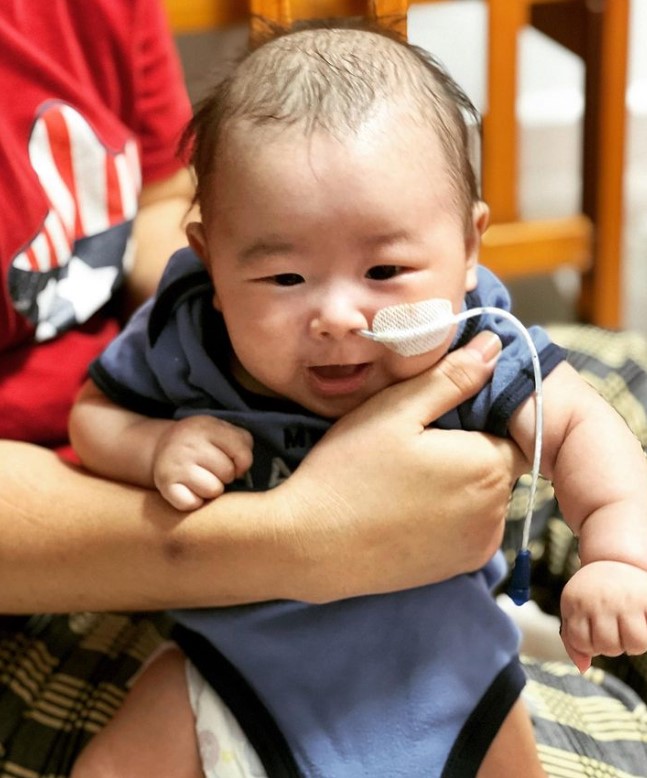 Photo from hope_for_baby_lucas IG
Photo from hope_for_baby_lucas IG
終完成治療並出院
對此,醫生馬上為王彥喆準備該藥物的注射與其他相關治療,在完成1小時的注射療程後效果也頗好。醫生表示雖然接下來的2至3個月需要進行密切的監督,尤其是藥物對於肝臟、心臟與血小板的影響,但目前王彥喆亦已經完成治療並康復,能夠回家休養。另外,其母親綺恩也表示「我們好興奮,想借此機會,告訴所有關心我們的朋友,彥喆完成注射了!感謝大家的幫忙和關懷」。
什麽是「脊髓性肌肉萎縮症」?
「脊髓性肌肉萎縮症」(Spinal Muscular Atrophy,SMA)是一種罕見的神經肌肉系統疾病,通常是由一個或多個遺傳突變所引起的。這種疾病主要影響脊髓中的運動神經細胞,這些神經細胞負責控制肌肉的運動。當這些神經細胞受損或死亡時,肌肉無法正常運作,導致肌肉萎縮和虛弱。
然而「脊髓性肌肉萎縮症」的嚴重程度也可以因患者的基因而有所不同,有幾種不同的SMA亞型:
1. SMA I:被稱為嬰兒期脊髓性肌肉萎縮症,是最嚴重的亞型,患者通常會在出生不久後就出現無法坐立或行走的症狀。
2. SMA II:被稱為兒童期脊髓性肌肉萎縮症,這種亞型的症狀會在幼兒時期開始出現,患者可以學會坐立,但無法行走。
3. SMA III:被稱為青少年或成人期脊髓性肌肉萎縮症,這是最輕度的亞型,症狀通常會在兒童或青少年時期開始,患者能夠行走一段時間,但會在日後慢慢變得虛弱。
Text : UrbanLife Health Editorial
Photos : UrbanLife Health Editorial、hope_for_baby_lucas IG